

Abteilung für Dermatologie, St. John's Medical College and Hospital, Koramangala, Bangalore, Karnataka-560 034, Indien
Korrespondenzadresse :
Fiona F Sequeira
Abteilung für Dermatologie, St. John's Medical College and Hospital, Koramangala, Bangalore, Karnataka-560 034
Indien
|
So zitieren Sie diesen Artikel: Sequeira FF, Jayaseelan E. Keratosis follicularis spinulosa decalvans bei einer Frau. Indian J Dermatol Venereol Leprol 2011;77:325-327 |
Copyright: (C)2011 Indian Journal of Dermatology, Venereology, and Leprology
Abstrakt
Keratosis follicularis spinulosa decalvans (KFSD) ist ein seltenes follikuläres Syndrom, das mit ausgedehnter Keratosis pilaris und fortschreitender vernarbender Alopezie einhergeht. Diese Genodermatose beginnt oft im Säuglingsalter oder in der frühen Kindheit und wird X-chromosomal vererbt. Vorwiegend sind Jungen betroffen, Mädchen zeigen häufig keine oder nur eine milde Form der Erkrankung. Wir beschreiben diese nicht so häufige Form von KFSD bei einem neunjährigen Mädchen.
Schlüsselwörter: Keratosis follicularis spinulosa decalvans, weiblich, Kind
Einführung
Keratosis pilaris atrophicans (KPA) ist der Oberbegriff für eine Gruppe von drei seltenen und unterschiedlichen klinischen Einheiten, die die vernarbenden Typen der Keratosis pilaris darstellen. Drei Kategorien von KPA sind: Keratosis pilaris atrophicans faciei (KPAF), Atrophoderma vermiculatum (AV) und Keratosis follicularis spinulosa decalvans (KFSD). Sie haben die folgenden Merkmale gemeinsam: keratotische follikuläre Papeln, nichteitrige Entzündungen unterschiedlichen Ausmaßes und atrophische Endstadien, die durch irreversiblen Haarausfall und/oder atrophische Vertiefungen ähnlich wie Narben gekennzeichnet sind. Alle diese Erkrankungen scheinen erblich bedingt zu sein. Obwohl sie in ihrer charakteristischen Form als harmlose Erkrankung gilt, kann die Läsion in vielen Fällen kosmetische Probleme verursachen. Es gibt keine wirksame Therapie.
Fallbericht
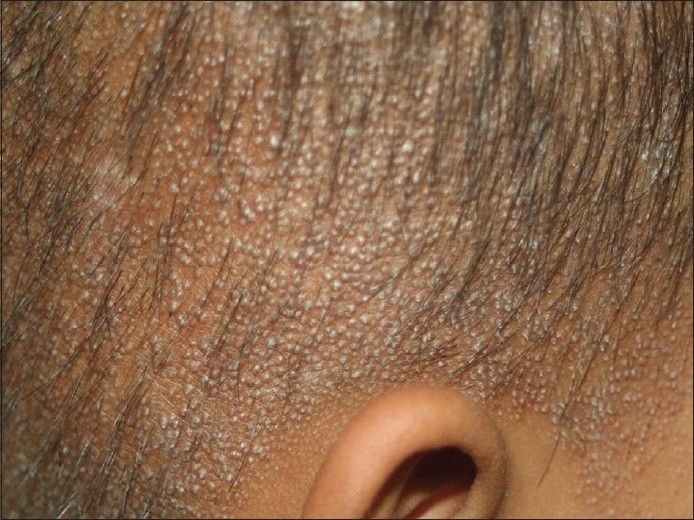
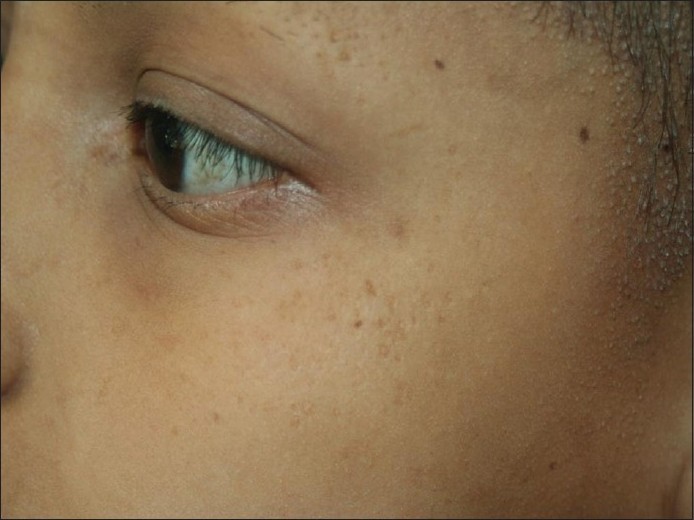
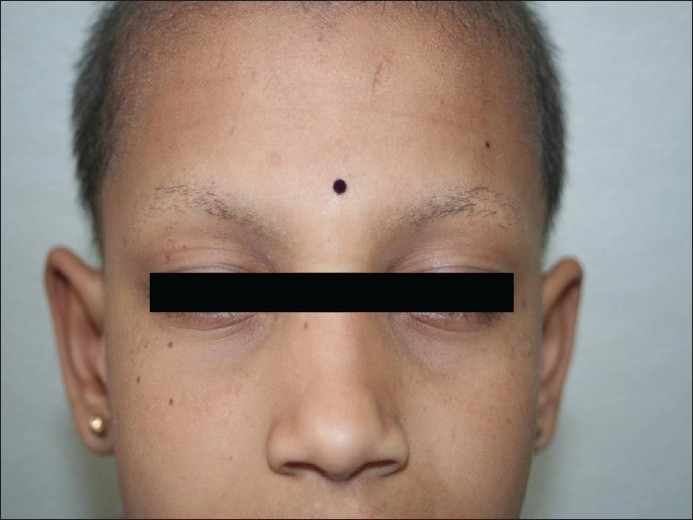
Ein neunjähriges Mädchen aus einer blutsverwandten Ehe ersten Grades kam in unsere Ambulanz und klagte über raue Haut auf der Kopfhaut seit dem fünften Lebensjahr und am ganzen Körper seit dem achten Lebensjahr in Verbindung mit teilweisem Verlust von Kopf- und Augenbrauenhaar. Bei der Geburt bemerkten ihre Eltern das Fehlen von Kopf- und Augenbrauenhaar, das im Laufe der nächsten drei bis vier Jahre allmählich bis zu einem gewissen Grad nachwuchs und schließlich spärlich wurde. Die Familienanamnese war nicht ursächlich. Bei der körperlichen Untersuchung wurden mehrere follikuläre fleischfarbene Hornpapeln auf der Kopfhaut, den Augenbrauen, den Wangen und den oberen und unteren Gliedmaßen festgestellt. Bei näherer Betrachtung der Kopfhaut, der Wangen und der Augenbrauen wurden feine Schuppen und Bereiche mit vernarbender Alopezie, punktförmiger Atrophie und Haarausfall in der seitlichen Hälfte der Augenbrauen festgestellt [Abbildung - 1], [Abbildung - 2], [Abbildung - 3]. Zähne, Nägel, Handflächen und Fußsohlen waren normal. Sie hatte keine Photophobie in der Anamnese. Die ophthalmische Untersuchung ergab keine abnormalen Befunde
 |
 |
 |
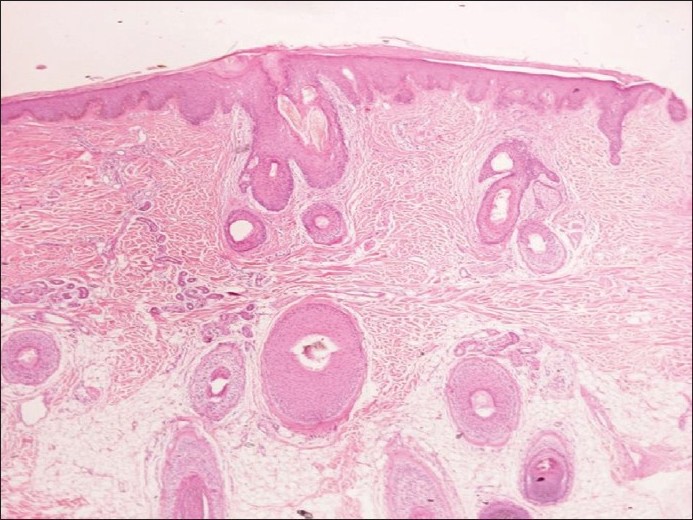
Um Monilethrix auszuschließen, wurde eine Haarmikroskopie durchgeführt, die in diesem Fall normal war. Eine Stanzbiopsieprobe von der Kopfhaut zeigte eine Follikelverstopfung in der Epidermis mit leichter Akanthose und frühe perifollikuläre Fibrose mit leichtem lymphozytischem Infiltrat. Die Dermis war dünner geworden und wies unauffällige, missgebildete Talgdrüseneinheiten auf. Die Haarschäfte schienen normal zu sein [Abbildung 4]. Aufgrund aller oben genannten Befunde wurde die klinische Diagnose Keratosis follicularis spinulosa decalvans gestellt. Angesichts der Infektion, die sie infolge der Exkoriation hatte, wurde bei unserer Patientin fünf Tage lang Azithromycin oral verabreicht, außerdem jeden zweiten Tag abends topische 0,025 % Retinsäure für die Kopfhaut und tagsüber eine Salicylsäure-Steroid-Lotion. Der Körper wurde mit Feuchtigkeitscremes mit Harnstoff und Lecithin behandelt. Einen Monat später zeigte sich bei der Patientin eine deutliche Besserung im Sinne einer verringerten Rauheit, eines verstärkten Haarwuchses und eines ausbleibenden Krankheitsfortschritts. Sie wurde gebeten, die gleiche topische Behandlung fortzusetzen und in einem Monat zur Untersuchung wiederzukommen.
 |
Diskussion
Keratosis follicularis spinulosa decalvans, eine relativ seltene Erkrankung, wurde erstmals von Macleod beschrieben, [1] allerdings war es Siemens, der diesen beschreibenden Begriff erstmals im Jahr 1926 verwendete. [2] Der Vererbungsmodus ist X-chromosomal und einem Locus bei Xp 22.13 – p 22.2 zugeordnet. [3] Die Erkrankung wurde jedoch auch in Familien beobachtet, bei denen das Übertragungsmuster auf einen autosomal-dominanten Erbgang hindeutet. [4] Einige Autoren verwendeten die Bezeichnung Folliculitis spinulosa decalvans (FSD), um die autosomal-dominante Form zu definieren. Diese lässt sich von der häufigeren X-chromosomalen Form durch eine stärker ausgeprägte Follikelentzündung, die besonders auf der Kopfhaut sichtbar ist, unterscheiden. [5] Im Allgemeinen sind Männer schwerer betroffen, während heterozygote Frauen in Familien mit X-chromosomalem dominantem Erbgang einen milderen Phänotyp aufweisen können. [6] Der Prozess der Lyonisierung (nicht zufällige X-Inaktivierung) könnte die Expression von KFSD bei Frauen erklären. Eine Studie von Aten et al . zeigte, dass KFSD-Patienten Mutationen im MBTPS2-Gen tragen und dass dies die normale Funktion des Proteins beeinträchtigt, indem die MBTPS2-Aktivität verringert wird. MBTPS2 wird für die Spaltung von sterolregulatorischen Element-bindenden Proteinen benötigt. [7]
Die Erkrankung beginnt im Kindesalter mit zahlreichen Hornpfropfen und Milien auf Nase und Wangen und später auf Augenbrauen, Kopfhaut, Hals und Körper. Vernarbende Alopezie auf Kopfhaut, Augenbrauen und Wimpern wird im Kindesalter sichtbar und schreitet bis zur Pubertät fort. Zu den damit verbundenen Merkmalen gehören Palmoplantarkeratose, mit Vorliebe für die Fersenregion, und das ungewöhnliche Zeichen hoher Nagelhaut (oder langer Nagelhaut). [6] Zu den Augenanomalien gehören Photophobie, Keratitis, Konjunktivitis, angeborenes Glaukom und Linsenstar. [2] Weniger häufig berichtete Merkmale sind Atopie, Taubheit, geistige Behinderung, Akne keloidalis nuchae und Haarfollikulitis, Aminoazidurie und wolliges Haar. [8], [9], [10], [11]
KFSD simuliert das Ichthyosis follicularis Alopecia Photophobia (IFAP)-Syndrom. Letzteres ist durch nicht vernarbende Alopezie, ausgedehnte KP, schwere Photophobie und Hornhautdystrophie gekennzeichnet. [12], [13] Das Vorliegen einer vernarbenden Alopezie bei unseren Patienten spricht für die Diagnose KFSD gegenüber dem IFAP-Syndrom. Die anderen Follikelerkrankungen, von denen es unterschieden werden muss, sind Lichen planopilaris und Lichen spinulosus. Ersterer ist durch atrophische oder vernarbende Flecken auf der Kopfhaut mit vollständigem Verlust der Follikelöffnungen gekennzeichnet. Die umgebenden marginalen Haarfollikel und verbleibenden kleinen Haarinseln innerhalb des Flecks weisen perifollikuläre erythematöse Flecken und Schuppen auf. An den Rändern des sich ausbreitenden Alopeziebereichs sind häufig akuminierte keratotische Pfropfchen zu beobachten. [14] Letztere ist eine seltene und gutartige Erkrankung, die durch follikuläre keratotische Papeln gekennzeichnet ist, die Hals, Gesäß, Bauch, Trochanterregionen, Knie und Streckseiten der Arme betreffen. Lichen spinulosus hat eine Vorliebe für akrale Bereiche, im Gegensatz zur Keratosis pilaris, die häufig auf die oberen Bereiche der Arme und Beine beschränkt ist. Der für Lichen spinulosus charakteristische Hornstachel kann entfernt werden, wobei eine winzige trichterförmige Öffnung in der Papel zurückbleibt, während bei Keratosis pilaris eine ganze einzelne Läsion mit dem Pfropf entfernt werden kann. [15]
Bisher ist keine wirksame Therapie für KFSD bekannt. Die häufige Anwendung topischer Keratolytika und Erweichungsmittel verbessert die Hautstruktur. [4] Antibiotika wie Tetracycline, Sulfonamide (Dapson), Makrolide, Penicilline und Rifampin wurden in therapeutischen Dosen verwendet und als unwirksam befunden. [4], [16] Topische und intraläsionale Kortikosteroide wurden versucht, führten jedoch zu vorübergehender Besserung. Etretinat und Isotretinoin wurden ebenfalls verwendet, jedoch mit unterschiedlichen Ergebnissen. [4], [17] Es ist wahrscheinlich, dass Retinoide, die bei Verhornungsstörungen nützlich sind, wirken, indem sie den Prozess der follikulären Hyperkeratose und Entzündung herunterregulieren. Die laserunterstützte Haarentfernung mit dem langpulsigen, nicht-gütegeschalteten Rubinlaser hat sich bei progressiver oder hartnäckiger KFSD als nützlich erwiesen. [18]
Verweise
| 1. | Macleod JM. Drei Fälle von Ichthyosis follicularis in Verbindung mit Kahlheit. Br J Dermatol 1909;21:165-89. [Google Scholar] |
| 2. | Rand R, Baden HP. Keratosis follicularis spinulosa decalvans – Bericht über zwei Fälle und Literaturübersicht. Arch Dermatol 1983;199:22-6. [Google Scholar] |
| 3. | Porteous ME, Strain L, Logie LJ, Herd RM, Benton EC. Keratosis follicularis spinulosa decalvans: Bestätigung der Verbindung mit Xp22.13-p22.2. J Med Genet 1998;35:336-7. [Google Scholar] |
| 4. | Baden HP, Byers HR. Klinische Befunde, Hautpathologie und Therapieansprechen bei 21 Patienten mit Keratosis pilaris atrophicans. Arch Dermatol 1994;130:469-75. [Google Scholar] |
| 5. | Oranje AP, Van Osch LD, Oosterwijk JC. Keratosis pilaris atrophicans. Arch Dermatol 1994;130:500-2. [Google Scholar] |
| 6. | Van Osch LD, Oranje AP, Keukens FM, van Voorst Vader PC, Veldman E. Keratosis follicularis spinulosa decalvans: Eine Familienstudie mit sieben männlichen Fällen und sechs weiblichen Trägern. J Med Genet 1992;29:36-40. [Google Scholar] |
| 7. | Aten E, Brasz LC, Bornholdt D, Hooijkaas IB, Porteous ME, Sybert VP, et al . Keratosis follicularis spinulosa decalvans wird durch Mutationen in MBTPS2 verursacht. Hum Mutat 2010;31:1125-33. [Google Scholar] |
| 8. | Britton H, Lustig J, Thompson BJ, Meyer S, Esterly NB. Keratosis follicularis spinulosa decalvans: Ein Säugling mit Gedeihstörungen, Taubheit und wiederkehrenden Infektionen. Arch Dermatol 1978;114:761-4. [Google Scholar] |
| 9. | Grosshans E, Heid E, Stoll C. Keratosis follicularis spinulosa decalvans und Aminosäuren. Ann Dermatol Venereol 1978;105:433-8. [Google Scholar] |
| 10. | Lacarrubba F, Dall'Oglio F, Rossi A, Schwartz RA, Micali G. Familiäre Keratosis follicularis spinulosa decalvans in Verbindung mit wolligem Haar. Int J Dermatol 2007;46:840-3. [Google Scholar] |
| 11. | Janjua SA, Iftikhar N, Pastar Z, Hosler GA. Keratosis follicularis spinulosa decalvans in Verbindung mit Akne keloidalis nuchae und Haarfollikulitis. Am J Clin Dermatol 2008;9:137-40. [Google Scholar] |
| 12. | Herd RM, Benton EC. Keratosis follicularis spinulosa decalvans: Bericht über eine neue Abstammung. Br J Dermatol 1996;134:138-42. [Google Scholar] |
| 13. | Eramo LR, Esterly NB, Zieserl EJ, Stock EL, Herrmann J. Ichthyosis follicularis mit Alopezie und Photophobie. Arch Dermatol 1985;121:1167-74. [Google Scholar] |
| 14. | Mehregan DA, Van Hale HM, Muller SA. Lichen planopilaris: Klinische und pathologische Studie an 45 Patienten. J Am Acad Dermatol 1992;27:935-42. [Google Scholar] |
| 15. | Boyd AS. Lichen spinulosus: Fallbericht und Übersicht. Cutis 1989;43:557-60. [Google Scholar] |
| 16. | Bellet JS, Kaplan AL, Selim MA, Olsen EA. Keratosis follicularis spinulosa decalvans in einer Familie. J Am Acad Dermatol 2008;58:499-502. [Google Scholar] |
| 17. | Alfadley A, Al Hawsawi K, Hainau B, Al Aboud K. Zwei Brüder mit Keratosis follicularis spinulosa decalvans. J Am Acad Dermatol 2002;47:S275-8. [Google Scholar] |
| 18. | Chui CT, Berger TG, Price VH, Zachary CB. Behandlung hartnäckiger vernarbender Follikelerkrankungen durch laserunterstützte Haarentfernung: Ein vorläufiger Bericht. Dermatol Surg 1999;25:34-7. [Google Scholar] |





































